摘要
馮希佩爾-林道綜合征是一種罕見的遺傳性常染色體顯性疾病。其病程伴有多種腫瘤的發展,以下腫瘤最常見於中樞神經係統的血管母細胞瘤、透明細胞腎細胞癌、嗜鉻細胞瘤、胰島腫瘤和內淋巴囊腫瘤。此外,腎髒和胰腺囊腺瘤和附睾囊腺瘤已被診斷為男性,子宮寬韌帶囊腺瘤已被診斷為女性。
本文介紹一個以視力障礙為首發症狀的男孩的診斷方法。在眼科會診期間觀察到高血壓性視網膜病變和極高的血壓。超聲心動圖確診高血壓並發症,診斷高血壓心肌病。經光學相幹斷層掃描證實為高血壓性視網膜病變。神經學、心髒病學和內分泌學檢查顯示雙側嗜鉻細胞瘤是動脈高血壓的原因。此外,一些遺傳學研究顯示VHL ex1 p.Y112 C基因突變導致嗜鉻細胞瘤的遺傳形式,證實了von Hippel-Lindau綜合征。在嗜鉻細胞瘤手術治療後,患者需要根據馮希佩爾-林道病的監測方案仔細管理。
背景
Von Hippel-Lindau綜合征(視網膜-小腦血管瘤病)是一種罕見的,基於遺傳的,常染色體顯性疾病。它的過程伴隨著多種腫瘤的發展,以下腫瘤最常見於中樞神經係統血管母細胞瘤、透明細胞腎細胞癌、嗜鉻細胞瘤、胰島腫瘤和內淋巴囊腫瘤。此外,男性診斷為腎、胰腺囊腺瘤和附睾囊腺瘤,女性診斷為子宮寬韌帶囊腺瘤[1- - - - - -3.].
該綜合症的發病率估計約為活產嬰兒的1/36 000 [4].這種疾病與位於3號染色體短臂的vhl基因的兩個等位基因突變有關。到目前為止,已經確定了超過150種導致該綜合征發展的突變[5- - - - - -11].
VHL病是一種以多發血管腫瘤為特征的遺傳性癌症綜合征。該綜合征是由von Hippel-Lindau蛋白(pVHL)失活引起的[6].功能VHL蛋白的缺失導致高水平的HIF,從而導致VEGF、PDGF和TGF- α的產生增加。這解釋了微血管細胞的生長和增殖。HIF還有助於嗜鉻細胞瘤中甲狀腺氨酸羥化酶和兒茶酚胺的過量生產。它是抑製神經脊細胞凋亡、嗜鉻細胞瘤和副神經節瘤發生的原因[1,6,7].該綜合征根據嗜鉻細胞瘤發展的風險分為兩種類型。
von Hippel-Lindau綜合征的譜包括Chuvash多血細胞血症,也稱為家族性2型紅細胞血症。這是一種與腫瘤無關的罕見疾病,在居住在伏爾加河地區的Chuvash人群中發病率特別高[1,6].
與VHL綜合征相關的腫瘤通常為雙側和多灶性,但很少惡性[2,7,12- - - - - -16].與散發性腫瘤不同的是,它們影響年輕患者[14].
這種疾病的最初症狀通常與視網膜、小腦和中樞神經係統其他部位的血管母細胞瘤有關[17].患者會出現虛弱、四肢疼痛、背痛、頭痛、麻木、頭暈等症狀。通常診斷為紅細胞生成素分泌過多引起的紅細胞增多症。
MRI用於診斷血管母細胞瘤。患有VHL綜合征10年以上的患者應每年進行一次MRI檢查。小而無症狀的腫瘤隻需要仔細觀察。位於小腦或視神經附近的有症狀的腫瘤以及大的血管母細胞瘤應通過神經外科手術切除或使用伽瑪刀治療。血管母細胞瘤和術後並發症是VHL患者身體殘疾的主要原因。
視網膜母細胞瘤發生於胎兒早期和<10歲。大多數患者沒有任何症狀。視網膜血管母細胞瘤可通過眼科檢查或熒光血管造影進行診斷。治療的選擇包括在疾病早期進行激光光凝治療。在與視力喪失相關的腫瘤中,應考慮切除受影響的眼睛。
視力缺陷的另一個原因是視網膜病變與嗜鉻細胞瘤發展過程中的動脈高血壓有關[18- - - - - -20.].在VHL綜合征的過程中,嗜鉻細胞瘤最常發生在兒童和年輕患者中。它們可以位於腎上腺和副神經節[14,15].嗜鉻細胞瘤最常見的症狀包括出汗、頭痛、焦慮、躁動和心悸,以及皮膚蒼白,這通常被認為是一種病理症狀[7,16- - - - - -25].根據一些作者的說法,陣發性或慢性動脈高血壓是嗜鉻細胞瘤的基本症狀。本課題的文獻資料顯示,兒童的動脈高血壓通常是慢性的,不像成年人,他們患有周期性的高血壓發作[24,25].長期高度的動脈緊張可導致並發症的發生,例如高血壓心肌病或視網膜病變伴視網膜脫離[26].散發性和固有嗜鉻細胞瘤的診斷是基於生化和影像學分析。激素活性腫瘤可通過測量血清或24小時尿液收集中兒茶酚胺和甲氧基兒茶酚胺的水平來檢測(可選擇的方法)[21].這種方法的高敏感性與腫瘤定期分泌兒茶酚胺(腎上腺素和去甲腎上腺素)有關,隻有在大量分泌這種物質的發作時才能檢測到兒茶酚胺的升高水平。相反,由於兒茶酚胺在腫瘤內部轉化為甲氧基兒茶酚胺,甲氧基兒茶酚胺的水平永久升高[27- - - - - -41].VHL綜合征過程中的嗜鉻細胞瘤被認為主要分泌去甲腎上腺素和甲氧去甲腎上腺素。反過來,在MEN 2或NF1綜合征過程中發生的散發性嗜鉻細胞瘤和腫瘤會分泌腎上腺素、去甲腎上腺素及其代謝產物[36,42- - - - - -44].實驗室檢查常顯示碳水化合物代謝紊亂、白細胞增多和低鈣血症。後一種情況可由腫瘤內鈣離子的增加隔離引起,腫瘤利用鈣離子同時釋放兒茶酚胺[44].腹腔鏡下腎上腺部分切除術,通常涉及糖皮質激素替代,是治療兒童嗜鉻細胞瘤的首選方法[32].
VHL綜合征除結節性病變外,分布在不同部位的囊性性病變也可發生。兩例附睾囊腺瘤作為VHL綜合征的首發症狀已在本學科文獻中報道[34],但胰腺和腎髒囊腺瘤也可能發生[15,30.].這些類型的病變,特別是複合囊腺瘤,需要嚴密的監護,因為有可能出現透明細胞腎細胞癌成分的實性腫塊[15,31].
在診斷VHL綜合征成分的情況下,應對患者及其家庭成員進行基因檢測。資格可根據麻省總醫院指定的標準[33].
基因測試應在以下地點進行:
- 1 /
任何患有兩種vhl相關病變的個體:HB、RCC、Pheo、內淋巴囊腫瘤、附睾或附件乳頭狀囊腺瘤、胰腺囊腺瘤和神經內分泌腫瘤。
- 2/
任何患有以下一種或多種疾病的人:中樞神經HB、嗜酸性粒細胞瘤或副神經節瘤、內淋巴囊瘤和附睾乳頭狀囊腺瘤。
- 3 /
任何診斷<20年的>型RCC患者,雙側或多發性RCC,多發性胰腺漿液性囊腺瘤和神經內分泌腫瘤,胰腺囊腫和任何VHL相關病變。VHL綜合征的分類如圖所示。1.
圖1 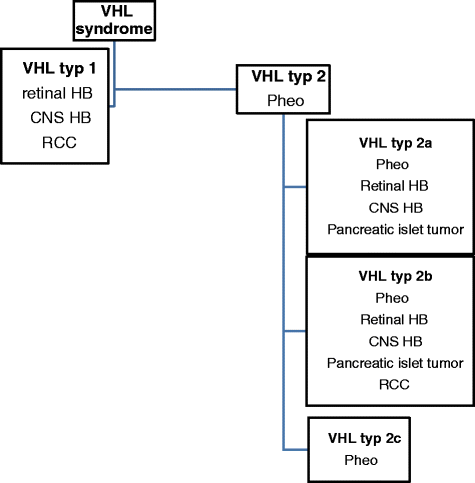
VHL綜合征的分類

案例展示
患者男,14歲,無複雜圍產期史(妊娠1次,分娩1次,38/39 hbd,體重3400克,身高54厘米,Apgar評分9分),因雙側盲點行眼科檢查。眼部檢查顯示眼壓升高和視網膜脫離區域,合並動脈高血壓升高(180/98 mmHg)提示高血壓性視網膜病變(圖2)。2).由於周期性頭痛和頭暈,患者被轉到神經內科作進一步診斷。入院時病情較好,體格檢查未發現神經係統綜合征征象;然而,動脈高血壓增加約。診斷為180/135 mmHg。頭部ct造影劑掃描顯示顳葉前部有3.5 × 2.5 × 4.0 cm的液體致密無造影劑增強區,鄰近骨板薄;圖像顯示的是蛛網膜囊腫。一個病灶位於右側顳葉皮層下(1cm),兩個病灶位於左右小腦半球(最大1.4 cm)。根據神經外科會診,這種現象需要觀察。雙側視神經在腔內及交叉部分未見病理征象。 The 3, 8 × 2 cm arachnoid cyst previously shown by CT located near the anterior pole of the right frontal lobe was also confirmed.

光學相幹斷層掃描顯示14歲男孩高血壓性視網膜病變。可見黃斑水腫和視網膜出血
由於動脈高壓持續在非常高的值(194/147 mmHg, 189/153 mmHg)和心動過速約120/min。發生後,患者轉診到兒科心內科,病情相當嚴重。超聲心動圖示高血壓心肌病征象,二尖瓣II級反流、左室肥厚,無流出道梗阻。患者在住院期間病情急劇惡化,男童出現嚴重頭痛伴嘔吐、舌麻、左上下肢感覺異常。因懷疑高血壓危象期間中樞神經係統出血,立即行頭部CT掃描,顯示腦水腫征象。降壓藥和減充血劑,如卡托普利、氨氯地平、卡維地洛、速尿和20%甘露醇100ml靜脈注射。腹部超聲示右側腎上腺約41 × 35mm低回聲組織病變,彩色及功率多普勒未見血管化,左側腎上腺未見病理病變。主動脈和腎動脈多普勒超聲未見明顯異常。為確認典型嗜鉻細胞瘤症狀,再次采集患者病史;它提供的信息,病人患有周期性發作的焦慮,伴隨著皮膚蒼白和過度出汗。 Ultrasound examination results combined with the clinical manifestations suggested a phaeochromocytoma in the right adrenal gland; therefore, the patient was referred to the Department of Paediatric Endocrinology and Diabetology for further diagnosis and treatment. Laboratory tests revealed an elevated glucose level (143 mg/dl), hypokalemia (3,19 mmol/l), and hypocalcaemia (8,2 mg/dl). The 4-fold assays of catecholamine levels in blood serum showed a very high noradrenaline content accompanied by a normal level of adrenaline; in 24-hour urine collection, the catecholamine and methoxycatecholamine levels were substantially elevated (Table1).此外,測量神經元特異性烯醇化酶和降鈣素水平,並進行甲狀腺超聲檢查以排除成神經細胞瘤和MEN 2。這些檢查結果正常。影像學診斷包括額外的腹部CT掃描,確認右側腎上腺存在約50 × ap 40 × cc 52mm的約rl結節性病變,左側腎上腺存在類似但較小的約24mm直徑的病變(圖2)。3.).

腹部計算機斷層掃描顯示左右腎上腺雙側結節性病變(嗜鉻細胞瘤)
病變在動脈期明顯。此外,在右腎上部發現一個直徑約15毫米的液體密集囊性區。,在內分泌科維持降壓治療;患者臨床情況好轉,動脈張力穩定,令人滿意。在普盧默溶液預用藥後,患者由131年,我腎上腺素能受體的mibg同位素閃爍。48和72 h後進行全身檢查,靜脈注射放射性同位素72 h後使用腹部SPECT顯像。示蹤劑積累增加的區域在兩個腎上腺均可見,但右側腎上腺的麵積更大。經過充分的診斷和預用藥後,患者在外科接受了側腹膜雙側腎上腺切除術。
右側腎上腺組織病理學檢查見直徑5cm的實性囊性腫瘤,切麵呈灰櫻桃紅色,局限於腎上腺。
此外,另一個直徑1.5 cm的實體瘤被定位。除此之外,腎上腺沒有變化。顯微圖像:嗜鉻細胞瘤;免疫組化:嗜鉻粒蛋白A(+),突觸性素(+);增殖指數Ki67 3 ~ 4%;acc。到PASS係統:1 pt(膠囊侵入)。左腎上腺:直徑2.7 cm的包膜腫瘤,切麵呈灰櫻桃紅色,局限於腎上腺。顯微圖像:嗜鉻細胞瘤;免疫組化:增殖指數Ki67: < 2%; acc.to the PASS system: 0 pt.
患者一直在接受氫化可的鬆替代治療。
由於懷疑在病人的嗜鉻細胞瘤的遺傳形式,進行了基因測試。結果顯示VHL ex1 p.Y112 C基因發生突變,與嗜鉻細胞瘤的遺傳形式有關,並證實了von Hippel-Lindau綜合征。進一步的基因檢測規定,以檢測遺傳綜合征與嗜鉻細胞瘤成分的其他家庭成員。
目前,手術後20個月,患者感覺良好,沒有報告任何令人擔憂的症狀。在家中測量動脈張力和心率顯示結果正常。隨訪腹部及甲狀腺超聲檢查正常;術後8個月進行同位素分析(131 -我MIBG閃爍成像)未顯示任何示蹤劑異常積聚的區域。對照組24小時尿液收集中兒茶酚胺和甲氧基兒茶酚胺水平正常(表2)1).眼科檢查顯示上述病變明顯消退。患者視力明顯改善,無盲點征象。隨後的中樞神經係統MRI檢查除蛛網膜囊腫外未發現任何異常。以前檢測到的病變可能是由發展中的腦病引起的。左心室輕度肌肉肥大持續存在。
結論
全國健康和營養調查的最新數據顯示,10%的兒童和青少年患有高血壓前期,4%患有高血壓[41,45- - - - - -46].嗜鉻細胞瘤在持續高血壓患者中約占0.05%至0.1%。嗜鉻細胞瘤是一種罕見的青少年高血壓的次要原因,通常發生在10歲以上。兒童高血壓早期診斷的建議包括在每次就診時對所有3歲以上兒童進行高血壓篩查,並對所有被診斷為高血壓的兒童進行實驗室評估、超聲心動圖和腎血管成像[44].
高達20%的嗜鉻細胞瘤被診斷為兒童。大多數為功能性腫瘤,臨床表現為兒茶酚胺分泌亢進和腫瘤腫塊效應。越來越多的嗜鉻細胞瘤在有遺傳綜合征的兒童的症狀前篩查中被發現:多發性內分泌瘤2型、von Hippel-Lindau病和副神經節瘤綜合征[36].
本病例研究顯示VHL綜合征的複雜性導致診斷困難。這種疾病的第一個明顯跡象是動脈高血壓,在預防性檢查時應由醫生診斷,否則可能導致並發症。VHL綜合征典型腫瘤的診斷,特別是兒科患者的診斷,需要對患者及其家庭成員進行更全麵的診斷和基因檢測,並對突變基因攜帶者進行密切監督。這種方法有助於腫瘤病變的早期診斷和治療。
同意
本病例報告及任何隨附圖像的發表均獲得患者的書麵知情同意。書麵同意書的副本可供本刊主編查閱。
縮寫
- VHL:
-
馮Hippel-Lindau
- Pheo:
-
嗜鉻細胞瘤
- 視網膜HB:
-
視網膜成血管細胞瘤
- 中樞神經係統Hb:
-
中樞神經係統血管母細胞瘤
- 碾壓混凝土:
-
透明細胞腎細胞癌
- 131 i - mibg:
-
131i -元碘苄基胍
- 10月:
-
光學相幹層析成像
參考文獻
Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau病:分子病理學基礎,臨床標準,基因檢測,腫瘤臨床特征及治療中華臨床醫學雜誌,2006;36(6):337-43。
Von Hippel E. Die anatomische Grundlage der Von beschriebenen ' selhr seltenen Erkrankung der Netzhaut '。Graefes Arch Ophthalmol. 1911; 79:350-77。
Lindau A. study en uber Kleinhirncysten。視網膜血管瘤病。病原微生物學報1926;增刊1:1-128。
勒斐伏爾M,福克斯WD。嗜鉻細胞瘤和副神經節瘤綜合征:遺傳學和治療更新。Curr Oncol, 2014; 21:8-17。
Martins R., João Bugalho M.副神經節瘤/嗜鉻細胞瘤:臨床導向的基因檢測。國際內分泌學雜誌2014卷,文章ID 794187http://dx.doi.org/10.1155/2014/7941872014年12月25日訪問。
馬希爾,伊澤留斯,葉茨,利特爾,本傑明,哈裏斯,等。馮希佩爾-林道病:一項基因研究。中華醫學雜誌,1991;28(7):443-7。
Maher ER, Hapmut PH N, Richard S. Von Hippel-Lindau疾病:臨床和科學綜述。《歐洲學報》2011;19:617-23。
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM,等。Von-Hippel-Lindau疾病。《柳葉刀》雜誌。2003;361:2059 - 67。
ben BU, Eng C, Olschewski M, Berger DP, Laubenberger J, Altehöfer C,等。VHL C . 505t > C突變具有較高的年齡相關外顯率,但沒有增加總死亡率。《中華醫學雜誌》2001;38:508-14。
bruch H, Kishida T, Glavac D, Chen F, Pausch F, Höfler H,等。von Hippel-Lindau (VHL)疾病合並嗜鉻細胞瘤在德國黑森林地區:創始人效應的證據。Hum Genet, 1995; 95:551-6。
Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM,等。北美、歐洲和日本家族中馮·希佩爾-林道病(VHL)基因的種係突變。胡姆穆塔特,1996;8:348-57。
李國強,李國強,李國強。《柳葉刀》雜誌。2004;363:1231-4。
Jimerez C, Cote G, Arnold A, Gagel RF。回顧:明顯散發的嗜鉻細胞瘤或副神經節瘤患者是否應該篩查遺傳性綜合征?中國臨床內分泌雜誌,2006;
Maher ER, Yates JRW, Ferguson-Smith MA。von Hippel- Lindau病、散發性小腦血管母細胞瘤和腎細胞癌2期突變模型的統計分析中華醫學雜誌,1990;27(5):311-4。
Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT,等。馮希佩爾-林道病的臨床特征和自然史。《醫學雜誌》1990;
Sharma P, Dhull VS, Bal C, Malhotra A, Kumar R. Von Hippel-Lindau綜合征:68ga - dotanoc PET-CT顯示全疾病譜。中國放射學報。2014;15(1):169-72。
王曉燕,王誌強,王誌強,等。von Hippel-Lindaudisease的眼部表現:臨床和分子研究。《眼科科學》2002;43:3067-74。
Shah V, Zlotcavitch L, Herro AM, Dubovy SR, Yehoshua Z, Lam BL.雙側乳頭病變為顯色征。與馮希佩爾-林道病有關臨床眼科學。2014;8:623-8。
I-Linn ZL,長四分衛。罕見的雙側視盤腫脹與黃斑星形之九歲女童。小兒眼斜視。2007;44(4):245-7。
Ba-Abbad RA, Nowilaty SR.雙側視盤腫脹為兒童嗜鉻細胞瘤的表現。中華醫學雜誌。2008;10(7):176。
嗜鉻細胞瘤的診斷和治療。波蘭動脈高血壓學會指南。2006;10(1):1 - 9。
李誌剛,李誌剛,李誌剛。嗜鉻細胞瘤-嗜鉻細胞瘤。波蘭內分泌。2006;57(1):54-62。
嗜鉻細胞瘤患者術前管理的探討。中國臨床內分泌雜誌,2007;29(4):489 - 497。
李誌強,李誌強,等。嗜鉻細胞瘤惡性標誌物的研究。波蘭內分泌學。2005;6:946-51。
Bissadaa NK, Safwata AS, Seyamc RM。兒童和青少年嗜鉻細胞瘤:臨床譜。中華兒科雜誌。2008;43:40 - 3。
Tibbetts MD, Wise R, Forbes B,等。由嗜鉻細胞瘤引起的兒童高血壓性視網膜病變:學校視力篩查失敗後的識別。J AAPOS。2012; 16:97-9。
李誌剛,李誌剛,李誌剛。嗜鉻細胞瘤致腎上腺素能性心肌炎的臨床研究。中華心血管醫學雜誌。2011;13:4。
Kopyta I, marzza E.兒童中風的危險因素。中風。2004;6(2):57 - 64。
Abourazzak S, Atmani S, Arqam LE, Chaouki S, Labib S, Harrandou M,等。缺血性腦卒中和雙側嗜鉻細胞瘤。BMJ Case rep 2010;2010:bcr12.2009.2535。
Beck O, Fassbender WJ, Beyer P, Kriener S, Neumann HP, Klingebiel T,等。兒童嗜鉻細胞瘤:進一步診斷的意義。兒科學報,2004;93:1630-4。
李誌強,李誌強,李誌強。嗜鉻細胞瘤。《柳葉刀》雜誌。2005;366:665 - 75。
Young Jr WF。內分泌高血壓。In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR,編輯。威廉斯內分泌學教科書。第11版。費城:Saunders Elsevier出版社;2008.505 - 37頁。
D'Herbomez M, Forzy G, Bauters C, Tierny C, Pigny P, Carnaille B,等。66例嗜鉻細胞瘤的生化診斷分析。中華內分泌雜誌,2007;56:569 - 75。
Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB,等。嗜鉻細胞瘤:第一屆國際研討會的臨床實踐建議。2005年10月。中華實用內分泌雜誌2007;3:92-102。
李誌強,李誌強,李誌強。嗜鉻細胞瘤生物化學篩選方法的研究進展。中國臨床內分泌雜誌,2004;
Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M,等。兒童嗜鉻細胞瘤和副神經節瘤的病因學、診斷和治療綜述。中國臨床內分泌雜誌,2010;35(5):529 - 529。
Eisenhofer G, lender JW, Linehan WM, Walther MM, Goldstein DS, Keiser HR。血漿去甲腎上腺素和甲腎上腺素檢測von Hippel-Lindau病和多發性內分泌瘤2型嗜鉻細胞瘤。中華醫學雜誌1999;34:1872 - 9。
Eisenhofer G, lender JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC,等。測定血漿甲氧基酪胺、去甲腎上腺素和甲腎上腺素作為不同遺傳性嗜鉻細胞瘤的鑒別指標。臨床化學雜誌,2011;57(3):411-20。
梁RS, Biswas SV, Duncan M, Rankin S. von Hippel-Lindau病的影像學特征。射線照相。2008;28:65 - 79。
mc侄女KL, Poffenbarger TS, Turner JL, Franco KD, Sorof JM, Portman RJ。青少年高血壓和高血壓前期患病率。中華兒科雜誌,2007;
Moore WE, Eichner JE, Cohn EM, Thompson DM, Kobza CE, Abbott KE。一個多種族學區的血壓篩查:健康兒童項目。《中國日報》2009;22:351-6。
李誌強,李誌強。嗜鉻細胞瘤。在:De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO,編輯。來源文本[互聯網]。南達特茅斯(馬薩諸塞州):MDText.com, Inc;2000.01訪問。2015.
劉誌剛,劉誌剛,劉誌剛,等。Von Hippel-Lindau綜合征:血管母細胞瘤和嗜鉻細胞瘤的診斷和治療。案例報告,2013;2013:624096。doi:10.1155 / 2013/624096.
Tootee A, Hasani-Ranjbar S. Von Hippel -Lindau病:一個老問題的新方法。中華內分泌雜誌,2012;10(4):619-24。
這些趨勢到底意味著什麼?循環。2007;116:1437-9。
兒童高血壓:新趨勢和挑戰。臨床科學(倫敦)。2010; 119:151 - 61。
作者信息
作者及隸屬關係
相應的作者
額外的信息
相互競爭的利益
作者宣稱他們之間沒有利益衝突。
作者的貢獻
KS在概念和設計,或數據的獲取,以及數據的分析和解釋方麵做出了重大貢獻;已經最終批準了將要出版的版本。BSI在概念和設計,或數據的獲取,以及數據的分析和解釋方麵做出了重大貢獻;曾參與撰寫稿件或對重要知識內容進行批判性修改;對即將出版的版本給予最後批準;並同意對工作的各個方麵負責,確保與工作的任何部分的準確性或完整性相關的問題得到適當的調查和解決。兩位作者都閱讀並批準了最終的手稿。
權利和權限
開放獲取本文根據創作共用屬性4.0國際許可協議(http://creativecommons.org/licenses/by/4.0/),允許在任何媒介上不受限製地使用、分發和複製,前提是您對原作者和來源給予適當的讚揚,提供到創作共用許可證的鏈接,並注明是否進行了更改。創作共用公共領域奉獻棄權書(http://creativecommons.org/publicdomain/zero/1.0/)除另有說明外,適用於本條所提供的資料。
關於本文

引用本文
Kozaczuk, S, Ben-Skowronek, I.從動脈高血壓並發症到von Hippel-Lindau綜合征診斷。兒科J醫院41, 56(2015)。https://doi.org/10.1186/s13052-015-0158-y
收到了:
接受:
發表:
DOI:https://doi.org/10.1186/s13052-015-0158-y
關鍵字
- 動脈高血壓
- 嗜鉻細胞瘤
- 馮希佩爾-林道綜合征